
MB PDX lines
Ex vivo drug treatments, eVLP transduction and tandem-mass-tag (TMT)-proteomic study on PDXs were performed at St. Jude Children’s Research Hospital (SJCRH). NSG mice (NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ; The Jackson Laboratory, JAX catalogue no. 005557) were used as hosts for PDX studies. Female NSG mice at least 8 weeks of age were anaesthetized in a surgical suite, and dissociated PDX cells were implanted in the cerebellum to amplify tumour material for downstream analyses. Mice were observed daily and euthanized at the onset of signs of sickness, including lethargy and neurological abnormalities. All clinical signs at the time of euthanasia did not exceed humane end point as determined by the SJCRH Institutional Animal Care and Use Committee (IACUC protocol no. 589-100536). RCMB51, RCMB52 and RCMB28 were originated and shared by R. J. Wechsler-Reya, Columbia University (previously Sanford Burnham Prebys). ICB1299 and ICB1572 were originated and shared by X.-N. Li, Northwestern University Feinberg School of Medicine (previously Baylor University). MED411FH, MED411FH-TC (established for tissue culture), MED211FH and MED2312FH were purchased from the Brain Tumor Research Laboratory, Seattle Children’s Hospital (previously Fred Hutchinson)44. Low passage PDXs (less than 10) were dissected and then flash-frozen for proteomics or dissociated for transduction and/or ex vivo drug sensitivity screening. Sample size choice was made to be at least n > 2 dissociated tumours for a given PDX model. No randomization of samples or blinding was conducted.
Sample processing of mouse PDX tissues for TMT mass spectrometry
Frozen tissues (20–30 mg) from each mouse PDX tumour were added to 200 ml of freshly prepared 8 M urea lysis buffer (containing 12 g of urea, 10X HEPES in 25 ml of Millipore ultrapure water) and homogenized with glass beads in a Bullet Blender Tissue Homogenizer (Next Advance) for 5 min, followed by a 2-min centrifugation at 2,000 rpm. Subsequently, 1% sodium deoxycholate was immediately added to the lysed tissues and vortexed for 2 min, followed by centrifugation at 1,000 rpm. The resulting supernatants were collected and stored at −80 °C. For quality control and quantification, 2 ml of lysates from each sample were electrophoresed on 4–12% NuPAGE gels (Invitrogen)45.
Protein digestion and TMT labelling
We performed the analysis with a previously optimized protocol45,46. For whole-proteome profiling, quantified protein samples (300 µg in the lysis buffer with 8 M urea) for each TMT channel were proteolysed with Lys-C (Wako, 1:100 w/w) at 21 °C for 2 h, and diluted by fourfold to reduce urea to 2 M for the addition of trypsin (Promega, 1:50 w/w) to continue the digestion at 21 °C overnight. The insoluble debris was kept in the lysates for the recovery of insoluble proteins. The digestion was terminated by the addition of 1% trifluoroacetic acid. After centrifugation, the supernatant was desalted with the Sep-Pak C18 cartridge (Waters), and then dried by Speedvac (Thermo Fisher). Each sample was resuspended in 50 mM HEPES (pH 8.5) for TMT labelling and then mixed equally, followed by desalting for the subsequent fractionation. For the whole-proteome analysis alone, 0.1 mg of protein per sample was used.
Extensive two-dimensional liquid chromatography–tandem mass spectrometry
The TMT-labelled samples were fractionated by offline basic pH reverse phase liquid chromatography (LC), and each of these fractions was analysed by the acidic pH reverse phase liquid chromatography–tandem mass spectrometry (LC–MS/MS)47,48. We performed a 160-min offline LC run at a flow rate of 400 µl min−1 on an XBridge C18 column (3.5-μm particle size, 4.6 mm × 25 cm, Waters; buffer A: 10 mM ammonium formate, pH 8.0; buffer B: 95% acetonitrile, 10 mM ammonium formate, pH 8.0)45. A total of 80 2-min fractions were collected. Every 41st fraction was concatenated into 40 pooled fractions, which were subsequently used for whole-proteome TMT analysis.
In the acidic pH LC–MS/MS analysis, each fraction from basic pH LC was dried by a Speedvac and was run sequentially on a column (75 µm × 35 cm for the whole proteome, 50 µm × 30 cm for whole proteome, 1.9 µm of C18 resin from Dr. Maisch, 65 °C to reduce backpressure) interfaced with a Fusion mass spectrometer (Thermo Fisher) for the whole proteome where peptides were eluted by a 90 min gradient (buffer A: 0.2% formic acid, 5% DMSO; buffer B: buffer A plus 65% acetonitrile). Mass spectrometry (MS) settings included the MS1 scan (450–1600 m/z, 60,000 resolution, 1 × 106 automatic gain control and 50-ms maximal ion time) and 20 data-dependent MS2 scans (fixed first mass of 120 m/z, 60,000 resolution, 1 × 105 automatic gain control, 110-ms maximal ion time, higher-energy collisional dissociation, 36% normalized collision energy, 1.0 m/z isolation window with 0.2 m/z offset and 10-s dynamic exclusion).
Protein identification and quantification with JUMP software
The computational processing of identification and quantification was performed with the JUMP search engine47. All original target protein sequences were reversed to generate a decoy database that was concatenated to the target database. Putative peptide spectrum matches (PSMs) were filtered by mass accuracy and then grouped by precursor ion charge state and filtered by JUMP-based matching scores (Jscore and ΔJn) to reduce false discovery rate (FDR) below 1% for proteins during the whole-proteome analysis. If one peptide could be generated from multiple homologous proteins, on the basis of the rule of parsimony, the peptide was assigned to the canonical protein form in the manually curated SwissProt database. If no canonical form was defined, the peptide was assigned to the protein with the highest PSM number. We performed the analysis in the following steps, as previously reported, with modifications49: (1) extracting TMT reporter ion intensities of each PSM; (2) correcting the raw intensities on the basis of the isotopic distribution of each labelling reagent (for example, TMT126 generates 91.8%, 7.9% and 0.3% of 126, 127 and 128 m/z ions, respectively); (3) excluding PSMs of very low intensities (for example, minimum intensity of 1,000 and median intensity of 5,000); (4) removing sample loading bias by normalization with the trimmed median intensity of all PSMs; (5) calculating the mean-centred intensities across samples (for example, relative intensities between each sample and the mean); (6) summarizing protein or phosphopeptide relative intensities by averaging related PSMs; (7) finally, deriving protein or phosphopeptide absolute intensities by multiplying the relative intensities by the grand-mean of three most highly abundant PSMs. In addition, we also performed y1 ion-based correction of TMT data. See Supplementary Data 1.
Analysis of differentially expressed proteins
Differentially expressed proteins were identified using an empirical Bayes-moderated t-test to compare treatment groups with the limma R package (v.3.54.2)50. Low expressions were defined as the lower 25th percentile of the means of the protein expression, and proteins with a prevalence of low expression in more than 70% of the samples were filtered out. As a result, 7,731 out of 11,428 proteins were retained for further analysis. Criteria for differential expression included a P value 51, with a confidence threshold greater than 0.7. The resulting networks were imported and visualized using Cytoscape (v.3.5.10). Interaction data were sourced from text mining, experiments and existing databases. See Supplementary Data 1 and 2.
Cell culture
HEK293T cells (Thermo Fisher) were a gift from B. E. Bernstein (Massachusetts General Hospital). Gesicle Producer 293T cells were a gift from D. R. Liu (Harvard University/Broad Institute) (Takara, catalogue no. 632617). K562 and CHLA-01-MED cells were obtained from ATCC. All mammalian cell lines were cultured in a humidified 5% CO2 incubator at 37 °C and routinely tested for mycoplasma (Sigma-Aldrich). ICB1299, CHLA-01-MED, and MED411FH-TC cells were cultured in stem cell media (50% DMEM/Nutrient Mixture F12 (DMEM/F12) plus 50% Neurobasal-A Medium supplemented with B-27 supplement (without vitamin A), 1 × GlutaMAX (Invitrogen), 1 mmol l−1 sodium pyruvate (Invitrogen), 1 × MEM Non-Essential Amino Acids Solution (Invitrogen), 25 mmol l−1 HEPES, 20 ng ml−1 basic fibroblast growth factor and 20 ng ml−1 epidermal growth factor). ICB1299 cells were cultured in Matrigel-coated plates and CHLA-01-MED and MED411FH-TC cells were cultured in low-attachment plates. HEK293F cells were obtained from Thermo Fisher. RPMI1640 and DMEM were supplemented with 100 U ml−1 penicillin and 100 µg ml−1 streptomycin (Gibco) and FBS (Peak Serum). K562 cells were cultured in RPMI1640 (Gibco) supplemented with 10% FBS. HEK293T and Gesicle Producer 293T cells were cultured in DMEM (Gibco) supplemented with 10% FBS. HEK293F cells were cultured in Freestyle 293 Expression Medium (Thermo Fisher) with shaking at 125 rpm. Spodoptera frugiperda (Sf9) insect cells (Expression Systems, catalogue no. 94-001F) were cultured in ESF921 media (Expression Systems) in a non-humidified and non-CO2 incubator at 27 °C with shaking at 140 rpm. High Five and ExpiSf9 cells were purchased from Thermo Fisher (catalogue nos. B85502 and A35243, respectively), with Grace insect medium (Thermo Fisher, catalogue no. 11595030) supplemented with 10% FBS (Cytiva) and 1% penicillin-streptomycin (Gibco), and cultured at 26 °C. All commercial cell lines were authenticated by short tandem repeat profiling (Genetica) and all cell lines were routinely tested for mycoplasma (Sigma-Aldrich).
Lentiviral production
For lentivirus production, transfer plasmids were co-transfected with GAG/POL and VSV-G plasmids into 293T cells using Lipofectamine 3000 (Thermo Fisher) according to the manufacturer’s protocol. Medium was exchanged after 6 h and the viral supernatant was collected 52 h after transfection and sterile-filtered (0.45 µm). K562 cells were transduced by spinfection at 1,800g for 1.5 h at 37 °C with 8 µg ml−1 polybrene (Santa Cruz). Where necessary, 48 h after transduction, cells were selected with 600 µg ml−1 G418 sulphate (Thermo Fisher).
Plasmid construction
Plasmids were cloned by Gibson Assembly using NEBuilder HiFi (New England Biolabs). Cloning strains used were NEB Stable (lentiviral) (New England Biolabs). Final constructs were validated by Sanger sequencing (Azenta/Genewiz).
All KBTBD4 expression plasmids encoded isoform 1 (human, residues 1–518) but longer isoform 2 (residues 1–534) numbering was used. CoREST expression plasmids encoded isoform 1 (human) full length (considered residues 4–485). Open reading frames (ORFs) of human KBTBD4 and CoREST (mammalian expression) were amplified from ORFs obtained from Horizon Discovery. The LSD1 ORF was a gift from R. Shiekhattar (University of Miami Miller School of Medicine). Full length HDAC1 ORF was a gift from E. Verdin (Addgene, catalogue no. 13820). The coding sequence of HDAC2 (amino acids 2–488) was synthesized by IDT. The coding sequence of full length NUDCD3 (human, residues 1–361) was synthesized by Twist Biosciences.
For transfection constructs, CoREST–FLAG and HA–KBTBD4 (WT or mutant) constructs were cloned into pcDNA3. For KBTBD4 overexpression constructs, KBTBD4 coding sequences were cloned into pSMAL mCherry, which was generated from pSMAL through introduction of an mCherry ORF into pSMAL (a gift from J. E. Dick, University of Toronto). For bacmid expression, KBTBD4 and NUDCD3 were cloned into pFastbac, a gift from T. Cech. For inducible expression constructs, KBTBD4 coding DNA sequence (CDS) was cloned into pInducer20 (Addgene, catalogue no. 44012). For eVLP constructs, sgRNA sequences were cloned into pU6-sgRNA (a gift from D. R. Liu, Harvard University/Broad Institute) by PCR amplification and co-transfected with pCMV-MMLVgag-3xNES-ABE8e (Addgene, catalogue no. 181751), pBS-CMV-gagpol (Addgene, catalogue no. 35614) and pCMV-VSV-G (Addgene, catalogue no. 8454), gifts from D. R. Liu, P. Salmon and B. Weinberg, respectively. eVLP sgRNA sequences are provided in Supplementary Table 1.
Production of eVLPs
eVLPs were produced as previously described22. In brief, Gesicle Producer 293T cells were seeded in T-75 flasks (Corning) at a density of 5 × 106 cells per flask. After 20–24 h, a mixture of plasmids expressing VSV-G (400 ng), MMLVgag–pro–pol (3,375 ng), MMLVgag–3xNES–ABE8e (1,125 ng) and an sgRNA (4,400 ng) were co-transfected into each T-75 flask using jetPRIME transfection reagent (Polyplus) according to the manufacturer’s protocols. At 40–48 h after transfection, producer cell supernatant was collected and centrifuged for 10 min at 4 °C and 2,000g to remove cell debris. The clarified eVLP-containing supernatant was filtered through a 0.45 μm PVDF filter (Sigma-Aldrich). The filtered supernatant was concentrated by ultracentrifugation using a cushion of 20% (w/v) sucrose (Sigma-Aldrich) in PBS. Ultracentrifugation was performed at 26,000 rpm for 2 h at 4 °C using an SW28 rotor in an Optima XE-90 Ultracentrifuge (Beckman Coulter). After ultracentrifugation, eVLP pellets were resuspended in cold PBS (pH 7.4). eVLPs were frozen and stored at −80 °C. eVLPs were thawed on ice immediately before use and repeated freeze–thaw was avoided.
eVLP transduction in cell culture
K562 cells were plated for transduction in 96-well plates (Cellstar Greiner Bio-one) at a density of 50,000 cells per well with 5 µg ml−1 polybrene (Santa Cruz) media. Base editor (BE)-eVLPs were added directly to the culture media in each well. Next, 50 µl of fresh medium was added after 6 h, and another 100 µl of media was added 48 h after transduction. Then, 72 h after transduction, cellular genomic DNA was isolated and genotyped as described below. Transduced cells were allowed to recover for 7–10 days before degradation assays were performed.
For cell viability assays, ICB1299, CHLA-01-MED and MED411FH-TC were transduced with eVLPs and cultured in Stem cell media. Cells were collected on day 3 for genotyping. Cell viability was measured on day 4 (reference) and day 11 (end point) for ICB1299 and CHLA-01-MED or on day 3 (reference) and day 10 (end point) for MED411FH-TC using Cell Titer-Glo Luminescent Cell Viability Assay 2.0 (Promega) with PHERAstar FSX microplate reader. End point readings were normalized to that of reference to determine relative growth during 7 days of culture. For immunoblotting, ICB1299 cells were transduced with eVLPs and cultured in Stem cell media for 5 days before collecting for immunoblotting or genotyping. Primers used for genotyping are provided in Supplementary Table 2.
Genotyping
Genomic DNA was extracted using QuickExtract DNA Extraction Solution (Biosearch Technologies) according to the manufacturer’s protocol. We subjected 100 ng of DNA to a first round of PCR (25–28 cycles, Q5 hot start high-fidelity DNA polymerase (New England Biolabs)) to amplify the locus of interest and attach common overhangs. Then, 1 µl of each PCR product was amplified in a second round of PCR (8 cycles) to attach barcoded adapters. Primer sequences are provided in Supplementary Tables 2 and 3. Final amplicons were purified by gel extraction (Zymo) and sequenced on an Illumina MiSeq. Data were processed using CRISPResso2 (ref. 52) using the following parameters: –quantification_window_size 20 –quantification_window_center -10 –plot_window_size 20 –exclude_bp_from_left 0 –exclude_bp_from_right 0 –min_average_read_quality 30 –n_processes 12 –base_editor_output.
CRISPR–Cas9-mediated genome editing
Knock-in of CoREST–GFP in K562 cells
mEGFP followed by a ‘GGGSGGGS’ linker was knocked into the C terminus of CoREST (that is, RCOR1) in K562 cells. sgRNA (sgRNA: TTCAAAGCCACCAGTTTCTC) targeting the C terminus of CoREST was cloned into a Cas9 plasmid, PX459 (ref. 53), and electroporated according to the manufacturer’s protocol (Neon Transfection System, Thermo Fisher) with a repair vector containing the mEGFP CDS and linker flanked by 750 base pairs of genomic homology sequences to either side of the CoREST C terminus. In brief, 2 × 105 cells were washed twice with PBS and resuspended in buffer R. PX459 (0.5 µg) and the repair vector (0.5 µg) were added to the cell suspension, and electroporated at 1,350 V with 10-ms pulse width for 4 pulses using the Neon Transfection System 10 µl kit. After electroporation, cells were immediately transferred to prewarmed media. To generate single-cell clones, cells were gated to sort for the top 0.2% GFP+ and single-cell sorted on a MoFlo Astrios EQ Cell Sorter (Beckman Coulter), and expanded and validated by western blot and Sanger sequencing.
Knock-in of HDAC2-dTAG in HDAC1-null CoREST–GFP K562 cells
Homology directed repair was used to insert a linker-FKBP12F36V-2xHA-P2A-PuroR cassette into the C terminus of HDAC2 in HDAC1-null CoREST–GFP K562 cells (generation described below). sgRNA (sgRNA: GGTGAGACTGTCAAATTCAG) (Synthego) targeting the C terminus of HDAC2 was electroporated according to the manufacturer’s protocol (Neon Transfection System, Thermo Fisher) with a repair vector containing the linker-FKBP12F36V-2xHA-P2A-PuroR CDS flanked by 700–800 base pairs of genomic homology sequences to either side of the HDAC2 C terminus. In brief, 2 × 106 cells were washed twice with PBS and resuspended in buffer R. The sgRNA and the repair vector (0.5 µg) were added to the cell suspension, and electroporated at 1,350 V with 10-ms pulse width for 3 pulses using the Neon Transfection System 100 µl kit. After electroporation, cells were immediately transferred to prewarmed media. After 9 days of recovery, cells were selected with 2 µg ml−1 puromycin (Thermo Fisher) for 10 days before single-cell sorting on a MoFlo Astrios EQ Cell Sorter (Beckman Coulter). Single-cell clones were validated by Sanger sequencing and western blot.
Generation of knockout K562s
HDAC1-null, HDAC2-null and KBTBD4-null CoREST–GFP K562 clones were generated using the Alt-R CRISPR–Cas9 System (IDT) to deliver ribonucleoprotein complexes containing knockout (KO) guides (HDAC1: GCACCGGGCAACGTTACGAA; HDAC2: TACAACAGATCGTGTAATGA; KBTBD4: GATATCTGTGAGTAAGCGGT) using the Neon Transfection System (Thermo Fisher) according to the manufacturer’s protocol. Transfected cells were recovered for 72 h before sorting for single-cell clones on a MoFlo Astrios Cell Sorter (Beckman Coulter). Single-cell clones were validated by genotyping and immunoblotting. For LSD1 knockout, lentiviral vectors carrying sgRNA (LSD1) were generated by cloning appropriate sequences (LSD1: TAGGGCAAGCTACCTTGTTA) into pLentiCRISPR.v2 lentiviral vector. Control vector contained sgRNA targeting luciferase (sgControl). Lentivirus was produced and K562 CoREST–GFP cells were transduced and puromycin selected as described above. Primers and guide sequences used for genotyping are provided in Supplementary Tables 3 and 4, respectively.
Degradation assay of KBTBD4 mutants
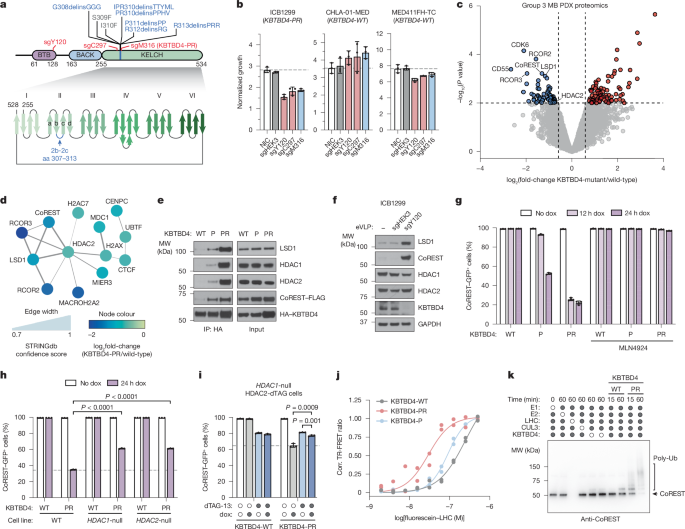
K562 KBTBD4-null CoREST–GFP cells were generated as described above. KBTBD4 overexpression constructs were cloned into pSMAL mCherry and point mutations were introduced into coding regions using standard PCR-based site-directed mutagenesis techniques. Lentiviral particles carrying the overexpression constructs were produced and used to transduce K562 KBTBD4-null CoREST–GFP cells as described above. At 48 h after transduction, GFP+ percentage was measured for mCherry+ cells in each condition (Supplementary Fig. 1a).
Inducible expression of KBTBD4 mutants
Lentiviral particles carrying the inducible constructs were produced and used to transduce K562 cells as described above. At 48 h after transduction, cells were selected with 600 µg ml−1 G418 for 8–10 days. The selected cells were then treated with 1 µg ml−1 dox for indicated times with or without pre-treatment of DMSO, MLN4924 (1 µM), SAHA (10 µM), CI-994 (10 µM) or RBC1HI (10 µM). GFP+ percentage was measured for cells in each condition as shown in Supplementary Fig. 1a.
Immunoblotting
Cells were lysed on ice in RIPA buffer (Boston BioProducts) with 1X Halt Protease Inhibitor Cocktail (Thermo Fisher) and 5 mM EDTA (Thermo Fisher). Lysate was clarified by centrifugation and total protein concentration was measured with the BCA Protein Assay (Thermo Fisher). Samples were electrophoresed and transferred to a 0.45-μm nitrocellulose membrane (Bio-Rad). Membranes were blocked with Tris-buffered saline Tween (TBST) with 5% Blotting-Grade Blocker (Bio-Rad) and incubated with primary antibody at the following dilutions: KBTBD4 (Novus Biologicals, catalogue no. NBP1-88587, 1:1,000), HDAC1 (Cell Signaling Technology, catalogue no. 34589, D5C6U, 1:1,000), HDAC2 (Cell Signaling Technology, catalogue no. 57156, D6S5P, 1:1,000), FLAG (Sigma-Aldrich, catalogue no. F1804, M2, 1:2,000), HA-tag (Cell Signaling Technology, catalogue no. 3724, C29F4, 1:1,000), GAPDH (Santa Cruz, catalogue no. sc-47724, 0411, 1:10,000). Membranes were washed three times with TBST and incubated with secondary antibody at the following dilutions: anti-rabbit IgG HRP conjugate (Promega, catalogue no. W4011, 1:20,000), anti-mouse IgG HRP conjugate (Promega, catalogue no. W4021, 1:40,000). Unless otherwise stated, following three washes with TBST, immunoblots were visualized using SuperSignal West Pico PLUS or SuperSignal West Femto chemiluminescent substrates (Thermo Fisher).
Co-immunoprecipitation
HEK293T cells were transfected with 2 µg of pcDNA3 HA-KBTBD4 plasmid (mutant or WT) and with or without 3 µg of pcDNA3 CoREST–FLAG (full length or truncated) using PEI MAX transfection reagent (Polysciences) according to the manufacturer’s protocol. At 48 h after transfection, cells were treated with 1 µM MLN4924 for 3 h then with 1 µM UM171, 10 µM SAHA or vehicle for 1 h, or with 10 µM CI-994 for 3 h. Cells were washed twice with cold PBS and flash-frozen. Cells were thawed and lysed on ice in lysis buffer (25 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% NP-40 alternative) supplemented with cOmplete, EDTA-free Protease Inhibitor Cocktail (Sigma-Aldrich), and the lysates were cleared. The protein concentration was quantified as above and diluted to 1 mg ml−1 in lysis buffer with 1 µM UM171 or DMSO. Supernatants were immunoprecipitated overnight at 4 °C with 25 µl of Pierce anti-HA magnetic beads (Thermo Fisher). Beads were washed six times with lysis buffer, eluted in SDS–PAGE loading buffer and carried forward to immunoblotting as described above.
Protein expression and purifications
Human recombinant KBTBD4 for biochemical and biophysical analyses was purified from Sf9 insect cells. Complementary DNAs for human KBTBD4 and NUDCD3 proteins were cloned into the pFastBac donor vector and the recombinant baculoviruses were constructed using the Bac-to-Bac protocol and reagents (Thermo Fisher). KBTBD4 MB mutations were introduced into coding regions using standard PCR-based site-directed mutagenesis techniques. All KBTBD4 constructs were tagged on the N terminus with 6×His cleavable by TEV protease. These plasmids were used to prepare separate baculoviruses according to standard protocols (Bac-to-Bac Baculovirus Expression System, Thermo Fisher). Detection of gp64 was used to determine baculovirus titre (Expression Systems). For expression, SF9 cells were grown to a density of 1–2 × 106 cells per millilitre and co-infected with NUDCD3 baculovirus at a multiplicity of infection of 2 and KBTBD4 baculovirus at a multiplicity of infection of 3.5. The cells were incubated for 72 h (27 °C, 120g), collected and then frozen with liquid nitrogen for future purification. Cells were resuspended in lysis buffer (50 mM Tris-HCl, pH 8.0 cold, 500 mM NaCl, 1 mM Tris(2-carboxyethyl)phosphine (TCEP), 10% glycerol, 15 mM imidazole) supplemented with 1% NP-40, 1 mM PMSF and Roche Complete Protease Inhibitor and sonicated. Lysate was clarified by centrifugation at 100,000g for 30 min and incubated with His60 Ni Superflow affinity resin (Takara). Resin was washed with lysis buffer containing a stepwise gradient of 15–50 mM imidazole, followed by elution using lysis buffer with 250 mM imidazole. Eluate was exchanged into storage buffer (50 mM Tris-HCl, pH 8.0 cold, 150 mM NaCl, 1 mM TCEP, 10% glycerol) using an Econo-Pac 10DG desalting column (Bio-Rad) and further purified by size exclusion chromatography using a Superdex 200 10/300 GL column (GE Healthcare). The purity of the recombinant protein was verified by SDS–PAGE and fractions with 90–95% purity were pooled and stored at −80 °C.
Recombinant human KBTBD4 used in cryo-EM structure determination was purified from Trichoplusia ni High Five insect cells. cDNAs for human KBTBD4 and NUDCD3 proteins were cloned into the pFastBac donor vector and the recombinant baculoviruses were constructed using the Bac-to-Bac protocol and reagents (Thermo Fisher). KBTBD4 constructs were tagged on the N terminus with 10×His and MBP tag cleavable by TEV protease. These plasmids were used to prepare separate baculoviruses according to standard protocols (Bac-to-Bac Baculovirus Expression System, Thermo Fisher). For expression, the monolayer High Five cells were grown to about 80% confluency and co-infected with NUDCD3 baculovirus. The cells were incubated for 72 h (26 °C), collected and then frozen with liquid nitrogen for future purification. Cells were resuspended in lysis buffer (50 mM Tris-HCl, pH 8.0 cold, 150 mM NaCl, 1 mM TCEP, 20 mM imidazole) supplemented with 1 mM PMSF, 10 µM leupeptin, 0.5 µM aproptinin and 1 µM pepstatin A and sonicated. Lysate was clarified by centrifugation at 100,000g for 30 min and incubated with amylose affinity resin (New England BioLabs). Resin was washed with lysis buffer, followed by elution using lysis buffer with 10 mM maltose. Eluate was cut with tobacco etch virus protease overnight, followed by the prepacked anion exchange column (GE Healthcare) to get rid of the protease, and further purified by size exclusion chromatography using a Superdex 200 10/300 GL column (GE Healthcare). The purity of the recombinant protein was verified by SDS–PAGE and fractions with 90–95% purity were pooled and stored at −80 °C.
Recombinant LSD1–CoREST–HDAC complex was composed of full length LSD1 (UniProt ID: O60341) or LSD1 (Δ77–86), full length HDAC1 (UniProt ID: Q13547) and N-terminally truncated CoREST (amino acids 86–485) (UniProt ID: Q9UKL0) or N-terminal Cys CoREST26. The pcDNA3 vector was used to create plasmids encoding the different proteins. The CoREST constructs contained an N-terminal (His)10(Flag)3 tag followed by a TEV protease cleavage site. The constructs for ternary complex were co-transfected into suspension-grow HEK293F cells (Thermo Fisher) with polyethylenimine (Sigma) and collected after 48 h. Cells were resuspended in lysis buffer (50 mM HEPES, pH 7.5, 100 mM KCl, 5% glycerol, 0.3% Triton X-100, 1X Roche EDTA-free Complete Protease Inhibitor cocktail) and sonicated. Lysate was clarified by centrifugation at 12,000g for 30 min and incubated with Anti-FLAG M2 affinity gel (Sigma). The affinity gel was washed twice with lysis buffer and twice with SEC buffer (50 mM HEPES, pH 7.5, 50 mM KCl, 0.5 mM TCEP) followed by the incubation with TEV protease overnight at 4 °C. The complex was further purified by size exclusion chromatography using a Superose 6 10/300 column (GE Healthcare). The purity of the complex was verified by SDS–PAGE and fractions with 90–95% purity were pooled and supplemented with 5% glycerol and stored at −80 °C.
Recombinant HDAC2–CoREST complex, composed of HDAC2 (amino acids 2–488) (UniProt ID: Q92769) and CoREST (amino acids 86–485), was purified from ExpiSf9 cells (Thermo Fisher). cDNAs for human HDAC2 and CoREST proteins were cloned into the pFastBac donor vector and the recombinant baculoviruses were constructed using the Bac-to-Bac protocol and reagents (Thermo Fisher). HDAC2 (amino acids 2–488) construct was tagged on the N terminus with SUMO tag, which can be cleaved in insect cells and with 6×His on the C terminus. CoREST (amino acids 86–485) was tagged with 10×His tag followed by an MBP tag on the N terminus. To improve the solubility of CoREST, six amino acids were mutated to the corresponding residues found in MIER2 (W172K F188C F191E V197A V201N F209K). These plasmids were used to prepare separate baculoviruses according to standard protocols (Bac-to-Bac Baculovirus Expression System, Thermo Fisher). For expression, the suspension ExpiSf9 cells were grown to about 5 × 106 cells per millilitre and co-infected with HDAC2 and CoREST baculovirus. The cells were incubated for 72 h (26 °C), collected and then frozen with liquid nitrogen for future purification. Cells were resuspended in lysis buffer (50 mM Tris-HCl, pH 8.0 cold, 300 mM NaCl, 5 mM MaCl, 15% glycerol, 1 mM TCEP, 20 mM imidazole) supplemented with 1 mM PMSF, 10 µM leupeptin, 0.5 µM aproptinin and 1 µM epstatin A and sonicated. Lysate was clarified by centrifugation at 100,000g for 30 min and incubated with nickel affinity resin (Thermo Fisher). Resin was washed with lysis buffer, followed by elution using lysis buffer with 200 mM imidazole. Eluate was applied to the prepacked anion exchange column (GE Healthcare) to get rid of the contaminants and further purified by size exclusion chromatography using a Superdex 200 10/300 GL column (GE Healthcare). The purity of the recombinant protein was verified by SDS–PAGE and fractions with 90–95% purity were pooled and stored at −80 °C.
Fluorescein labelling of LHC
The fluorescein labelling of the LSD1–CoREST–HDAC1 complex was purified as described above. A Cys point mutagenesis has been conducted next to the TEV protease cleavage site of N-terminally truncated CoREST for the ligation reaction with NHS-fluorescein54. A 2 mM NHS-fluorescein was incubated with 500 mM mercaptoethanesulfonate (MESNA) in the reaction buffer (100 mM HEPES, pH 7.5, 50 mM KCl, 1 mM TCEP) for 4 h at room temperature in the dark for transesterification. The LSD1–CoREST–HDAC1 complex purified by FLAG M2 affinity gel was washed with reaction buffer and incubated with TEV protease for 5 h at 4 °C. The complex was then mixed with 500 µl of the fluorescein/MESNA solution to make a final concentration of 0.5 mM fluorescein and 125 mM MESNA. The mixture was incubated for 48 h at 4 °C in the dark. The complex was desalted by a Zeba spin desalting column (7 kDa molecular weight cut-off) and further purified by size exclusion chromatography using a Superose 6 10/300 column (GE Healthcare). Fluorescein labelling efficiency was analysed by SDS–PAGE and fluorescence gel imaging (Amersham Typhoon FLA 9500, Cytiva). The purity of the complex was verified by SDS–PAGE and fractions with 90–95% purity were pooled and supplemented with 5% glycerol and stored at −80 °C.
TR-FRET measurements
Unless otherwise noted, experiments were performed in white, 384-well microtitre plates (Corning, catalogue no. 3572) in 30-μl assay volume, or white, 384-well low-volume microtitre plates (PerkinElmer, catalogue no. 6008280). TR-FRET measurements were acquired on a Tecan SPARK plate reader with SPARKCONTROL software v.2.1 (Tecan Group), with the following settings: 340/50-nm excitation, 490/10-nm (Tb) and 520/10-nm (FITC, AF488) emission, 100-μs delay, 400-μs integration. The 490/10-nm and 520/10-nm emission channels were acquired with a 50% mirror and a dichroic 510 mirror, respectively, using independently optimized detector gain settings unless specified otherwise. The TR-FRET ratio was taken as the 520/490-nm intensity ratio on a per-well basis.
Ternary complex measurements by TR-FRET
Titration of fluorescein-labelled LSD1–CoREST–HDAC complex
Recombinant WT (or mutant) 6×His–KBTBD4 (10 nM, 2×) and CoraFluor-1-labelled anti-6×His IgG (5 nM, 2×)29 were diluted into LHC buffer, with or without 10 μM UM171, and 5 μl added to wells of a white, 384-well low-volume microtitre plate (PerkinElmer, catalogue no. 6008280). Serial dilutions of fluorescein-labelled LSD1–CoREST–HDAC complex (1:2 titration, 10-point, cmax = 1,000 nM, 2×) were prepared in ligand buffer and 5 μl added to wells of the same plate (final volume 10 μl, final 6×His–KBTBD4 concentration 5 nM, final CoraFluor-1-labelled anti-6×His IgG concentration 2.5 nM, fluorescein-labelled LSD1–CoREST–HDAC complex cmax = 500 nM). The plate was allowed to equilibrate for 1 h at room temperature before TR-FRET measurements were taken. Data were background-corrected from wells containing no 6×His–KBTBD4. Prism 9 was used to fit the data to a four-parameter dose–response curve.
Titration of InsP6
Recombinant WT or mutant (P and PR) 6×His–KBTBD4 (40 nM), fluorescein-labelled LSD1–CoREST–HDAC complex (40 nM) and CoraFluor-1-labelled anti-6×His IgG (20 nM)29 were diluted into a one-to-one mixture of ligand buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM TCEP, 10% glycerol) and LHC buffer (20 mM HEPES, pH 7.5, 1 mM TCEP, 2 mg ml−1 BSA, 0.1% Tween-20) and 10 μl added to wells of a white, 384-well low-volume microtitre plate (PerkinElmer, catalogue no. 6008280). InsP6 was added in serial dilution (1:10 titration, 6-point, cmax = 100 μM) using a D300 digital dispenser (Hewlett-Packard), and allowed to equilibrate for 1 h at room temperature before TR-FRET measurements were taken. Data were background-corrected from wells containing no InsP6. Prism 9 was used to fit the data to a four-parameter dose–response curve.
Incubation with HDAC inhibitor or UM171
Fluorescein-labelled LSD1–CoREST–HDAC complex (100 nM) and CoraFluor-1-labelled anti-6×His IgG (20 nM)29 were diluted into a one-to-one mixture of ligand buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM TCEP, 10% glycerol) and LHC buffer (20 mM HEPES, pH 7.5, 1 mM TCEP, 2 mg ml−1 BSA, 0.1% Tween-20, 100 µM InsP6) and 10 μl added to wells of a white, 384-well low-volume microtitre plate (PerkinElmer, catalogue no. 6008280). HDACi (SAHA, CI-994, RBC1HI) (10 µM), UM171 (10 µM) or vehicle (DMSO) was added using a D300 digital dispenser (Hewlett-Packard), and allowed to equilibrate for 1 h at room temperature. Recombinant WT or mutant (P or PR) 6×His–KBTBD4 (100 nM) was then added using a D300 digital dispenser (Hewlett-Packard), and allowed to equilibrate for 1 h at room temperature before TR-FRET measurements were taken. Data were background-corrected from wells containing no 6×His–KBTBD4. Prism 9 was used plot the data.
In vitro ubiquitination assay
The ubiquitination assays were set up similarly to as previously reported55. Reactions were performed at 37 °C in a total volume of 20 µl. The reaction mixtures contained 5 mM ATP, 100 μM WT ubiquitin, 100 nM E1 protein, 2 μM E2 protein, 0.5 μM neddylated RBX1-CUL3, 0.5 µM WT or PR KBTBD4 (unless otherwise indicated), with 25 mM Tris-HCl (pH 7.5), 20 mM NaCl, 10 µM InsP6 and 2.5 mM MgCl2 as reaction buffer. Substrate LHC at 0.5 µM was preincubated with everything except E1 in the reaction mixture at 37 °C for 5 min before adding E1 to initiate the reaction. Reactions were quenched at the indicated time points by adding SDS loading buffer containing reducing agent β-mercaptoethanol. The reaction samples were resolved on SDS–PAGE gels and analysed by Colloidal Blue staining, western blots or Typhoon fluorescent imaging.
Deep mutational scan
The library of KBTBD4 mutants in the 7-amino acid region between Gly307 and Arg313 was designed to comprise all possible: (1) deletions, (2) 1-amino acid substitutions, (3) 2-amino acid substitutions of adjacent residues, (4) 1-amino acid insertions, (5) 2-amino acid insertions, (6) 3-amino acid insertions of GGG or GSG, and (7) 100 randomly scrambled WT sequences and the 2 remaining MB indels (PR311delinsPPHV, IPR310delinsTTYML). The 5′ and 3′ homology arms were added as well as forward and reverse barcodes for the different sub pools of mutations for downstream cloning. The final library was ordered from Twist Biosciences as a pooled oligo library with final lengths of single-stranded oligos ranging from 101 to 113 nucleotides (Supplementary Data 3). The Twist pool was resuspended in tris-EDTA to the concentration of 1 ng μl−1 and the sub pools were separated by PCR amplification of 22 cycles, using lsPCR1 primers listed in Supplementary Table 5 and using 1 ng of the Twist pool as template in each reaction. Each sub pool was further amplified with lsPCR2 primers in Supplementary Table 5 by PCR amplification of 10 cycles and the library pools were gel purified (Zymo Gel DNA Recovery Kit). Oligos corresponding to 2–6-amino acid deletions were ordered from Sigma-Aldrich and cloned separately from the Twist pool (Supplementary Data 3).
The mutational library was cloned into pSMAL mCherry using Gibson assembly. The backbone for the Gibson assembly was prepared by introducing a BamHI restriction site in place of residues Gly307 and Arg313 using primers in Supplementary Table 6. The backbone was digested with BamHI (NEB) and subsequently treated with Antarctic phosphatase (NEB) and the correct linearized backbone was isolated by gel electrophoresis and purified using Gel DNA Recovery Kit (Zymo). Then, 190 ng of linearized vector and 13.15 ng of each sub pool were used for each Gibson reaction of 80 µl using HIFI DNA Assembly Master Mix (NEB). The Gibson reaction was incubated for 1 h at 50 °C and DNA was isolated by isopropanol precipitation and transformed into Lucigen Endura Competent Cells according to the manufacturer’s protocol. Cells were recovered in Lucigen Endura Recovery Media for 1 h at 30 °C and later plated and grown overnight at 30 °C. Colonies were collected and the plasmid library was extracted using QIAGEN Plasmid Maxi Kit. Purified sub pools were then combined for the final library and sequence verified on an Illumina MiSeq as previously described.
Lentivirus was produced and titred by measuring cell counts after transduction and mCherry selection. K562 KBTBD4-null CoREST–GFP knock-in cells were transduced with library lentivirus at a multiplicity of infection less than 0.3 and, at day 3 after transduction, cells were sorted on a MoFlo Astrios Cell Sorter (Beckman Coulter), collecting the top 10% GFP− and mCherry+, GFP+ and mCherry+, and mCherry+ (GPF+/−) cells. Genomic DNA was isolated using the QIAamp DNA Blood Mini kit or QIAamp UCP DNA Micro kit, and mutation sequences were amplified using barcoded primers listed in Supplementary Table 7, purified by gel extraction and sequenced on an Illumina MiSeq as previously described. Sorting was performed in three reps and, at all steps, greater than 150× coverage of the library was maintained.
We analysed data using Python (v.3.9.12) with Biopython (v.1.78), Pandas (v.1.5.1) and NumPy (v.1.23.4). In brief, raw reads matching sequences in the mutational library from unsorted as well as sorted (GFP+ and GFP−) cells were counted. Counts were then processed by converting them to reads per million, adding a pseudocount of 1 and transforming them by log2. Enrichment of each variant in GFP+ and GFP− populations was quantified by subtracting the GFP+ and GFP− log2-transformed counts, respectively, by corresponding log2-transformed counts for unsorted cells and averaged across replicates (Supplementary Data 4). Heatmaps were generated using matplotlib (v.3.7.1).
Analysis of sequence motifs
Position probability matrices of the GFP+ and unsorted populations were constructed for each mutually exclusive category (single substitution, single insertion, double substitution and double insertion) by normalizing raw counts by the total read counts of each corresponding category, averaging across replicates and tallying the probability of every amino acid at each position. The information content, IC, of each position N was calculated according to Kullback–Leibler divergence, which is as follows:
$${\rm{IC}}(N)=P(N)\times {\log }_{2}\frac{P(N)}{{B}_{N}}$$
where P(N) is the position probability matrix of the GFP+ population for each mutational category, and the position probability matrix of the unsorted population was used as background frequencies BN. Logos were generated using Logomaker (v.0.8)56.
Ex vivo drug sensitivity screening in MB PDX cells
MB PDXs harbouring WT KBTBD4 (RCMB28 n = 3, MED411FH n = 2) or KBTBD4-PR mutant (ICB1572 n = 5) were used to assess sensitivity to the HDAC1/2 inhibitor RBC1HI. In brief, freshly resected PDX tumours were cut into small pieces, incubated for 30 min at 37 °C in papain solution (10 units per millilitre, Worthington, catalogue no. LS003126) containing N-acetyl-l-cysteine (160 μg ml−1, Sigma-Aldrich, catalogue no. A9165) and DNase I (12 μg ml−1, Sigma-Aldrich, catalogue no. DN25) and dissociated to single cells by gentle pipetting. Red blood cells in the tumour cell suspension were removed by incubating in RBC Lysis buffer (STEMCELL technologies, catalogue no. 07850) at 37 °C for 2 min, followed by rinsing in DPBS-BSA. Cells were filtered using a 40-µm strainer and counted, and viability assessed to be above 80%. Cells were plated at 1,000 cells per well in 384-well plates in Stem cell media. Serially diluted RBC1HI was immediately added at a final concentration of 40–0.006 µM to the plated cells, with DMSO as negative control, and incubated for 72 h. Cell viability at the end of incubation was measured using Cell Titer-Glo Luminescent Cell Viability Assay 2.0 (Promega) with PHERAstar FSX microplate reader. Raw values were converted to cell viabilities and data analysed using Prism 10 to generate dose–response curves and obtain half-maximum inhibitory concentration values57.
Ex vivo degradation assay in MB PDX cells
KBTBD4-PR mutant PDX (ICB1572) tumour was freshly isolated from mouse cerebellum, dissociated to a single-cell suspension and plated at 1 × 106 cells per well in a six-well plate in Stem cell media. Cells were immediately dosed with the HDAC1/2 inhibitor RBC1HI or the NEDD8-activating enzyme inhibitor MLN4924 and incubated at 95% humidity and 5% CO2. Cells were collected 24 h later and lysed in RIPA buffer, and immunoblotting was performed. Immunoblot images were captured using the LICOR Odyssey CLX Imaging system.
Cryo-EM sample preparation and data collection
To assemble the complexes of KBTBD4-PR/TTYML–LHC and KBTBD4-PR–HDAC2–CoREST for cryo-EM study, the individually isolated KBTBD4-mutant proteins and co-expressed LHC or HDAC2–CoREST complex were mixed in stoichiometric amounts with 100 mM InsP6 added and subsequently applied to the Superose 6 increase gel filtration column (Cytiva) in a buffer containing 40 mM HEPES, pH 7.5, 50 mM KCl, 100 mM InsP6 and 0.5 mM TCEP. The isolated complex was then crosslinked with 37.5 mM glutaraldehyde at room temperature for 6 min and the reaction quenched with 1 M Tris-HCl pH 8.0. The crosslinked sample was snap-frozen for future use.
To prepare grids for cryo-EM data collection, a QuantiFoil Au R0.6/1 grid (Electron Microscopy Sciences) was glow discharged for 30 s at 20 mA with a glow discharge cleaning system (PELCO easiGlow). Then, 3.0 μl of the purified and crosslinked KBTBD4-PR/TTYML–LHC complex at 0.7 mg ml−1 or KBTBD4-PR–HDAC2–CoREST complex at 0.5 mg ml−1 was applied to a freshly glow-discharged grid. After incubating in the chamber at 10 °C and 100% relative humidity, grids were blotted for 3 s with a blotting force of zero, then immediately plunge-frozen in liquid ethane using a Vitrobot Mark IV system (Thermo Fisher). Data collection of KBTBD4-PR–LHC and KBTBD4-PR–HDAC2–CoREST was carried out on an FEI Titan Glacios and Krios transmission electron microscope (Thermo Fisher) operated at 200 kV and 300 kV, respectively, at the Arnold and Mabel Beckman Cryo-EM Center of the University of Washington. An automation scheme was implemented using the SerialEM software using beam-image shift at a nominal magnification of 105 K, resulting in a physical pixel size of 0.84 Å. The images were acquired on a K3 camera direct detector. The dose rate was set to 10 e− Å−2 s−1, and the total dose of 50 electrons per Å2 for each image was fractionated into 99 electron-event representation frames. Data collection of KBTBD4-TTYML–LHC was carried out on a Krios transmission electron microscope (Thermo Fisher) operated at 300 kV at the HHMI Janelia Research Campus. An automation scheme was implemented using the SerialEM58 software using beam-image shift59 at a nominal magnification of 165 K, resulting a physical pixel size of 0.743 Å. The images were acquired on a Falcon 4i camera direct detector, with the slit width of Selectris X (Thermo Fisher) set to be 6 eV. The dose rate was set to 15.39 e− Å−2 s−1, and the total dose of 60 electrons per Å2 for each image was fractionated into 60 electron-event representation frames. Data were collected in four sessions with a defocus range of 0.8–1.5 μm. In total, 6,839 and 8,414 videos were collected for KBTBD4-PR–LHC and KBTBD4-TTYML–HC, respectively. For KBTBD4-PR–HDAC2–CoREST, data were collected in four sessions with a defocus range of 0.8–1.8 μm. In total, 11,263 videos were collected.
Image processing and 3D reconstruction
For all three complexes, videos were collected and imported into CryoSPARC60 followed by patch motion correction and patch the contrast transfer function (CTF) estimation. Micrographs were kept after filtering the micrographs with CTF parameters and manual inspection. Blob picker job in CryoSPARC was able to pick particles, which were further extracted and subjected to two-dimensional classification. After five rounds of cleaning by two-dimensional classification, particles were selected and subjected to ab initio reconstruction. Subsequently, all particles were used for heterogenous refinement. After one extra round of cleaning up by heterogenous refinement, particles from good reconstruction were selected to get re-extracted without Fourier cropping. Homogenous refinement and non-uniform refinement61 help achieve an overall final resolution. To optimize the map for the KELCH-repeat domain, a soft mask focused on the KELCH domains was applied to local refinement, ending up with a further improved resolution. Topaz picker was used to pick more particles for a second round ab initio construction and refinements to achieve further resolution improvement. More details about the data processing can be found in Extended Data Figs. 5–7.
Model building and refinement
The initial structural models of the KBTBD4 dimer, the HDAC1/2–CoREST–ELM–SANT1 complex, were predicted with AlphaFold-Multimer in Google ColabFold2 (ref. 62). The structural models of KBTBD4 BTB-BACK domain, KELCH-repeat domain and HDAC1–CoREST were separately fit into the cryo-EM map using UCSF ChimeraX-1.7 (rc2023.12.12)63. The resulting model was subsequently rebuilt in Coot (0.9.8.91)64 on the basis of the protein sequences and the electron microscopy density and was further improved by real-space refinement in PHENIX (1.20.1-4487-000)65,66. The structure figures were made using PyMOL67.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.




